Differential expression between experimental groups using scde
Prepare data
Remove spike-in features
The analysis starts with a matrix of read counts, filtered based on gene and cell requirements. In this case, let us use the raw numbers of reads mapped to each endogenous feature, recast as integer type:
table(isSpike(sce.norm, "ERCC"))##
## FALSE TRUE
## 8226 44sce.endo <- sce.norm[!isSpike(sce.norm, "ERCC"),]## [1] 8226 342Subset data by time point
Considering the demonstrated impact on the Time factor on gene expression profiles, and the fact that differential expression will not be assessed between time points, individual time points will be processed separately for the remainder of this scde analysis.
2h
sce.2h <- sce.endo[,sce.endo$Time == '2h']
dim(sce.2h)## [1] 8226 1194h
sce.4h <- sce.endo[,sce.endo$Time == '4h']
dim(sce.4h)## [1] 8226 1106h
sce.6h <- sce.endo[,sce.endo$Time == '6h']
dim(sce.6h)## [1] 8226 113Filter count matrices
The analysis starts with a matrix of read counts, filtered based on gene and cell requirements.
In this case, for each time point, let us retain only features detected with at least 10 counts in at least 10 cells of any of the five experimental groups, use the raw numbers of reads mapped to each endogenous feature, recast as integer type.
Let us first define a function to apply the detection cutoff:
filterCounts <- function(m, counts = 10, cells = 10){
apply(m, 1, function(e){
return(sum(e >= counts) >= cells)
})
}2h
sg.2h <- droplevels(sce.2h$Group)
keep.2h <- filterCounts(counts(sce.2h))
cd.2h <- counts(sce.2h)[keep.2h,]; storage.mode(cd.2h) <- 'integer'## [1] 7755 1194h
sg.4h <- droplevels(sce.4h$Group)
keep.4h <- filterCounts(counts(sce.4h))
cd.4h <- counts(sce.4h)[keep.4h,]; storage.mode(cd.4h) <- 'integer'## [1] 7411 1106h
sg.6h <- droplevels(sce.6h$Group)
keep.6h <- filterCounts(counts(sce.6h))
cd.6h <- counts(sce.6h)[keep.6h,]; storage.mode(cd.6h) <- 'integer'## [1] 7604 113Error models
Here, we fit the error models on which all subsequent calculations will rely. The fitting process relies on a subset of robust genes that are detected in multiple cross-cell comparisons. Here we supply the groups argument, so that the error models for each experimental group of cells are fit independently. If the groups argument is omitted, the models would be fit using a common set.
2h
o.ifm.2h <- scde.error.models(
counts = cd.2h, groups = sg.2h, n.cores = 4, verbose = 1
)Particularly poor cells may result in abnormal fits, most commonly showing negative corr.a, and should be removed.
valid.cells <- o.ifm.2h$corr.a > 0; table(valid.cells)## valid.cells
## TRUE
## 119o.ifm.2h <- o.ifm.2h[valid.cells, ]4h
o.ifm.4h <- scde.error.models(
counts = cd.4h, groups = sg.4h, n.cores = 4, verbose = 1
)Particularly poor cells may result in abnormal fits, most commonly showing negative corr.a, and should be removed.
valid.cells <- o.ifm.4h$corr.a > 0; table(valid.cells)## valid.cells
## TRUE
## 110o.ifm.4h <- o.ifm.4h[valid.cells, ]6h
o.ifm.6h <- scde.error.models(
counts = cd.6h, groups = sg.6h, n.cores = 4, verbose = 1
)Particularly poor cells may result in abnormal fits, most commonly showing negative corr.a, and should be removed.
valid.cells <- o.ifm.6h$corr.a > 0; table(valid.cells)## valid.cells
## TRUE
## 113o.ifm.6h <- o.ifm.6h[valid.cells, ]Reorder counts to match error models
The scde.error.models produces error models with cells reordered by experimental group. For clarity, let us prepare a SCESet and count matrix that match this order:
2h
sce.ifm.2h <- sce.2h[,rownames(o.ifm.2h)]
sg.ifm.2h <- droplevels(sce.ifm.2h$Group)
cd.ifm.2h <- counts(sce.ifm.2h)[rownames(cd.2h),]; storage.mode(cd.ifm.2h) <- 'integer'4h
sce.ifm.4h <- sce.4h[,rownames(o.ifm.4h)]
sg.ifm.4h <- droplevels(sce.ifm.4h$Group)
cd.ifm.4h <- counts(sce.ifm.4h)[rownames(cd.4h),]; storage.mode(cd.ifm.4h) <- 'integer'6h
sce.ifm.6h <- sce.6h[,rownames(o.ifm.6h)]
sg.ifm.6h <- droplevels(sce.ifm.6h$Group)
cd.ifm.6h <- counts(sce.ifm.6h)[rownames(cd.6h),]; storage.mode(cd.ifm.6h) <- 'integer'Prior distribution for gene expression magnitudes
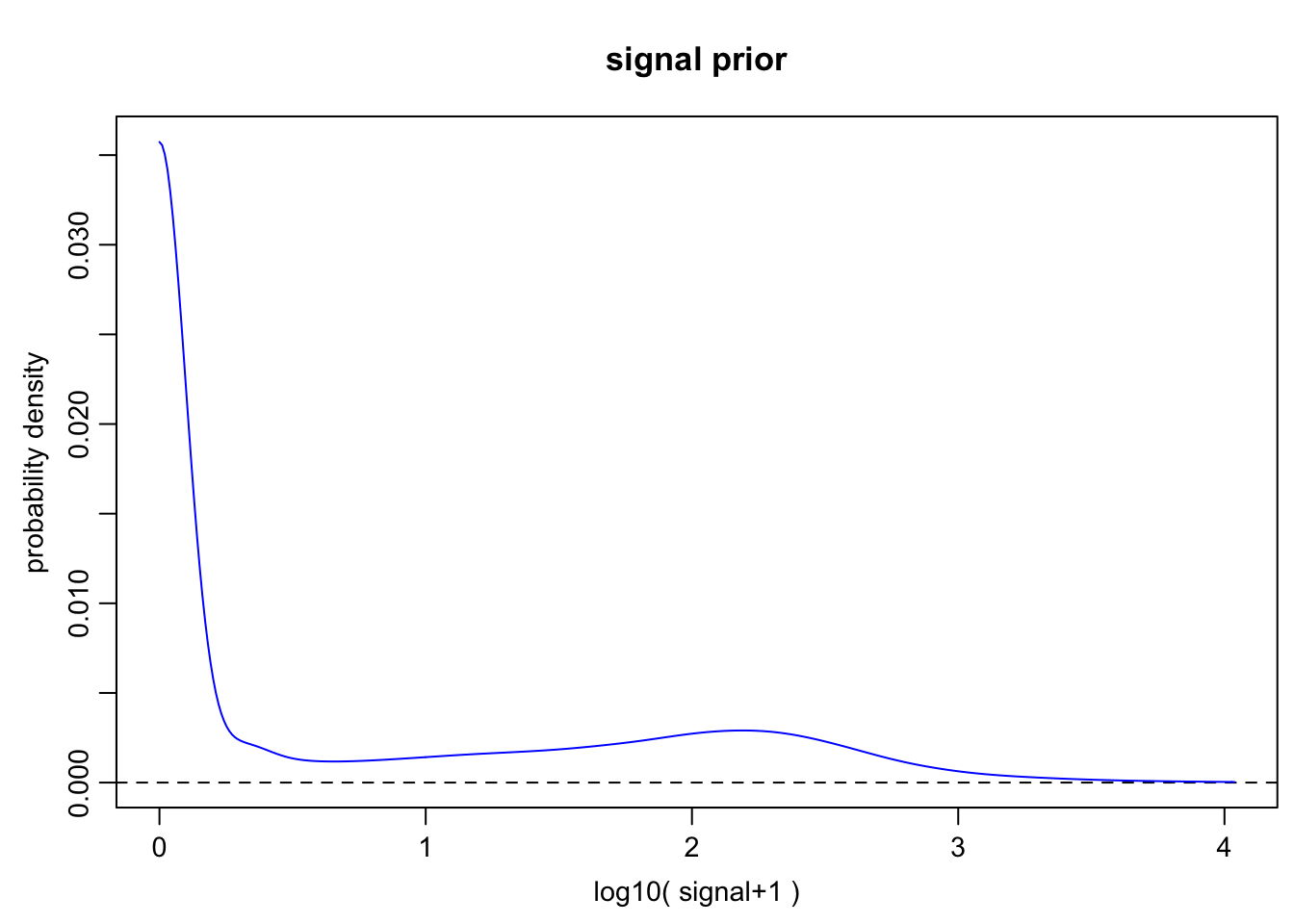
Finally, we need to define an expression magnitude prior for the genes. Its main function, however, is to define a grid of expression magnitude values on which the numerical calculations will be carried out.
2h
o.prior.2h <- scde.expression.prior(
models = o.ifm.2h, counts = cd.ifm.2h, show.plot = TRUE
)
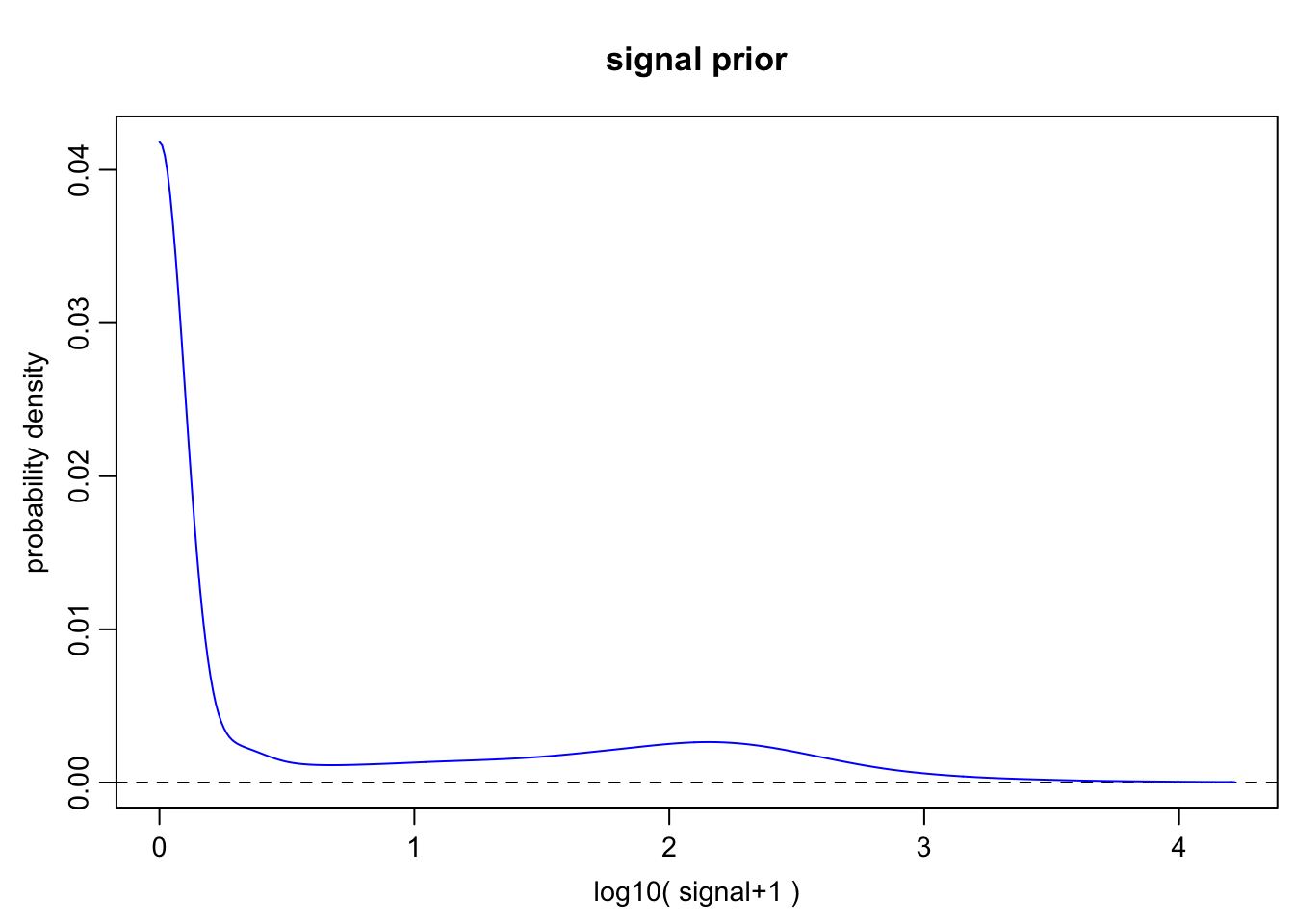
4h
o.prior.4h <- scde.expression.prior(
models = o.ifm.4h, counts = cd.ifm.4h, show.plot = TRUE
)
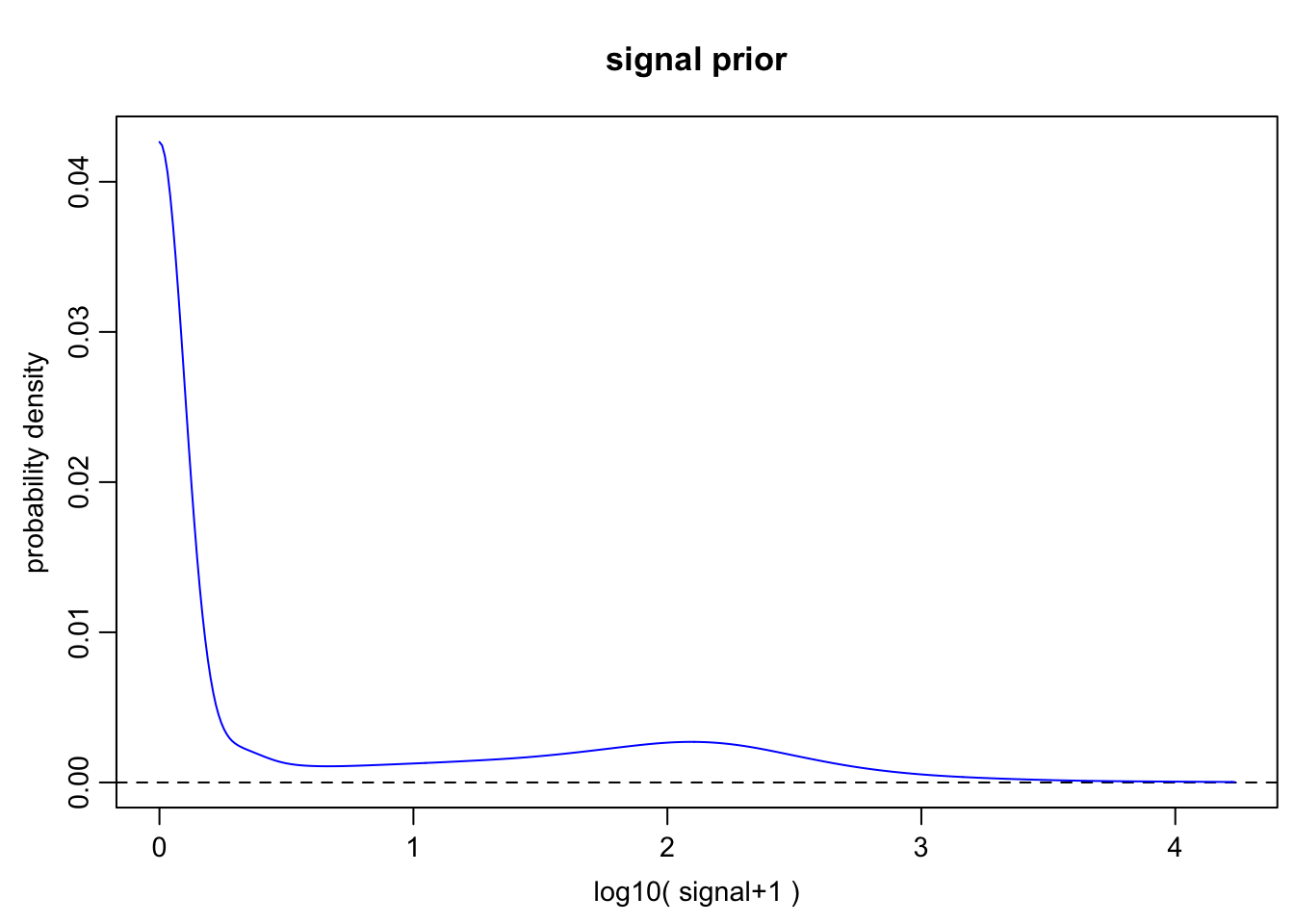
6h
o.prior.6h <- scde.expression.prior(
models = o.ifm.6h, counts = cd.ifm.6h, show.plot = TRUE
)
Differential expression
Setup
Let us first define:
- a list to store the result tables returned by scde
scde.res <- list()- a function used to convert the Z-score computed by scde to a empirical P-value (reference):
convert.z.score <- function(x, one.sided = NULL) {
z <- x$Z
if(is.null(one.sided)) {
pval = pnorm(-abs(z));
pval = 2 * pval
} else if(one.sided=="-") {
pval = pnorm(z);
} else {
pval = pnorm(-z);
}
x <- cbind(
x,
p.value = pval
)
return(x);
} - a function used to annotate the tables of results returned by scde:
addGeneName <- function(x){
x <- cbind(
gene_name = with(rowData(sce.endo), gene_name[match(rownames(x), gene_id)]),
x
)
return(x)
}- a function to order results by decreasing absolute Z-score:
orderResults <- function(x){
x <- x[with(x, order(abs(Z), decreasing = TRUE)),]
return(x)
}- a function to visualise scran-normalised gene expression for a given gene:
normExprsById <- function(geneId){
geneName <- subset(rowData(sce.endo),gene_id==geneId,"gene_name",drop=TRUE)
gdata <- data.frame(
logcounts = assay(sce.endo, "logcounts")[geneId,],
colData(sce.endo)[,c("Infection","Status","Time")],
row.names = colnames(sce.endo)
)
ggplot(gdata, aes(gsub("-", "\n", Infection), logcounts)) +
geom_violin(
aes(fill = Infection, alpha = Status),
draw_quantiles = c(0.25, 0.5, 0.75)
) +
geom_jitter(width = 0.1, alpha = 0.5, size = 0.5) +
facet_grid(Time ~ Status) +
scale_fill_manual(values = col.infection) +
scale_alpha_discrete("Status", range = c(0.3, 0.5)) +
ggtitle(sprintf("%s - %s", geneId, geneName)) +
theme_minimal() +
labs(y = "Normalised expression", x = "Infection") +
guides(alpha = "none")
}- functions to visualise scde estimate of expression and differential expression for a given gene (identifier) in a given contrast within a specific time point (code shown for 2h time point only):
single.scde.2h <- function(gene, groupTarget, groupRef){
gT <- sprintf("2h_%s", groupTarget); gR <- sprintf("2h_%s", groupRef)
sg.test <- factor(colData(sce.ifm.2h)[,"Group"], levels = c(gT, gR))
scde.test.gene.expression.difference(
gene, o.ifm.2h, cd.ifm.2h, o.prior.2h, sg.test,
n.cores = 4, verbose = 1
)
}- various significance levels:
sig.levels <- c(0.05, 0.01)
volcano.sig <- data.frame(
P = sig.levels,
level = as.character(sig.levels)
)- a function to visualise scde differential expression statistics, and return a table with those statistics augmented by a an empirical P value computed from the Z score returned by scde:
volcano.mle <- function(x, sub = NULL){
varName <- deparse(substitute(x))
x <- convert.z.score(x)
gg <- ggplot(x, aes(mle, -log10(p.value))) +
geom_point(aes(colour = (cZ != 0))) +
geom_hline(aes(yintercept=-log10(p.value),linetype=level),volcano.sig) +
ggtitle(varName, sub)
print(gg)
return(x)
}Contrasts
List
contrasts.2h <- list(
c("2h_STM-D23580_Infected", "2h_Mock_Uninfected"), # vs. Mock
c("2h_STM-LT2_Infected", "2h_Mock_Uninfected"),
c("2h_STM-D23580_Exposed", "2h_Mock_Uninfected"),
c("2h_STM-LT2_Exposed", "2h_Mock_Uninfected"),
c("2h_STM-D23580_Infected", "2h_STM-LT2_Infected"), # direct
c("2h_STM-D23580_Infected", "2h_STM-D23580_Exposed"),
c("2h_STM-LT2_Infected", "2h_STM-LT2_Exposed"),
c("2h_STM-D23580_Exposed", "2h_STM-LT2_Exposed")
)
contrasts.4h <- list(
c("4h_STM-D23580_Infected", "4h_Mock_Uninfected"), # 4h
c("4h_STM-LT2_Infected", "4h_Mock_Uninfected"),
c("4h_STM-D23580_Exposed", "4h_Mock_Uninfected"),
c("4h_STM-LT2_Exposed", "4h_Mock_Uninfected"),
c("4h_STM-D23580_Infected", "4h_STM-LT2_Infected"), # 4h
c("4h_STM-D23580_Infected", "4h_STM-D23580_Exposed"),
c("4h_STM-LT2_Infected", "4h_STM-LT2_Exposed"),
c("4h_STM-D23580_Exposed", "4h_STM-LT2_Exposed")
)
contrasts.6h <- list(
c("6h_STM-D23580_Infected", "6h_Mock_Uninfected"), # 6h
c("6h_STM-LT2_Infected", "6h_Mock_Uninfected"),
c("6h_STM-D23580_Exposed", "6h_Mock_Uninfected"),
c("6h_STM-LT2_Exposed", "6h_Mock_Uninfected"),
c("6h_STM-D23580_Infected", "6h_STM-LT2_Infected"), # 6h
c("6h_STM-D23580_Infected", "6h_STM-D23580_Exposed"),
c("6h_STM-LT2_Infected", "6h_STM-LT2_Exposed"),
c("6h_STM-D23580_Exposed", "6h_STM-LT2_Exposed")
)2h
stopifnot(all(rownames(o.ifm.2h) == colnames(cd.ifm.2h)))
for (contrastNames in contrasts.2h){
groupTarget <- contrastNames[1]; groupRef <- contrastNames[2]
sg.test <- factor(sce.ifm.2h$Group, levels = c(groupTarget, groupRef))
names(sg.test) <- colnames(sce.ifm.2h); summary(sg.test)
contrastName <- sprintf("%s-%s", groupTarget, groupRef); message(contrastName)
scde.res[[contrastName]] <- scde.expression.difference(
o.ifm.2h, cd.ifm.2h, o.prior.2h, sg.test, n.cores = 4, verbose = 1
)
}4h
stopifnot(all(rownames(o.ifm.4h) == colnames(cd.ifm.4h)))
for (contrastNames in contrasts.4h){
groupTarget <- contrastNames[1]; groupRef <- contrastNames[2]
sg.test <- factor(sce.ifm.4h$Group, levels = c(groupTarget, groupRef))
names(sg.test) <- colnames(sce.ifm.4h); summary(sg.test)
contrastName <- sprintf("%s-%s", groupTarget, groupRef); message(contrastName)
scde.res[[contrastName]] <- scde.expression.difference(
o.ifm.4h, cd.ifm.4h, o.prior.4h, sg.test, n.cores = 4, verbose = 1
)
}6h
stopifnot(all(rownames(o.ifm.6h) == colnames(cd.ifm.6h)))
for (contrastNames in contrasts.6h){
groupTarget <- contrastNames[1]; groupRef <- contrastNames[2]
sg.test <- factor(sce.ifm.6h$Group, levels = c(groupTarget, groupRef))
names(sg.test) <- colnames(sce.ifm.6h); summary(sg.test)
contrastName <- sprintf("%s-%s", groupTarget, groupRef); message(contrastName)
scde.res[[contrastName]] <- scde.expression.difference(
o.ifm.6h, cd.ifm.6h, o.prior.6h, sg.test, n.cores = 4, verbose = 1
)
}Volcano plots
Let us first identify the extreme values of maximum likelihood estimate of fold-change to scale subsequent plot for comparability and symmetry of axes:
mleRange <-
max(abs(do.call("c", lapply(scde.res, function(x){return(x$mle)}))))*c(-1,1)Uninfected
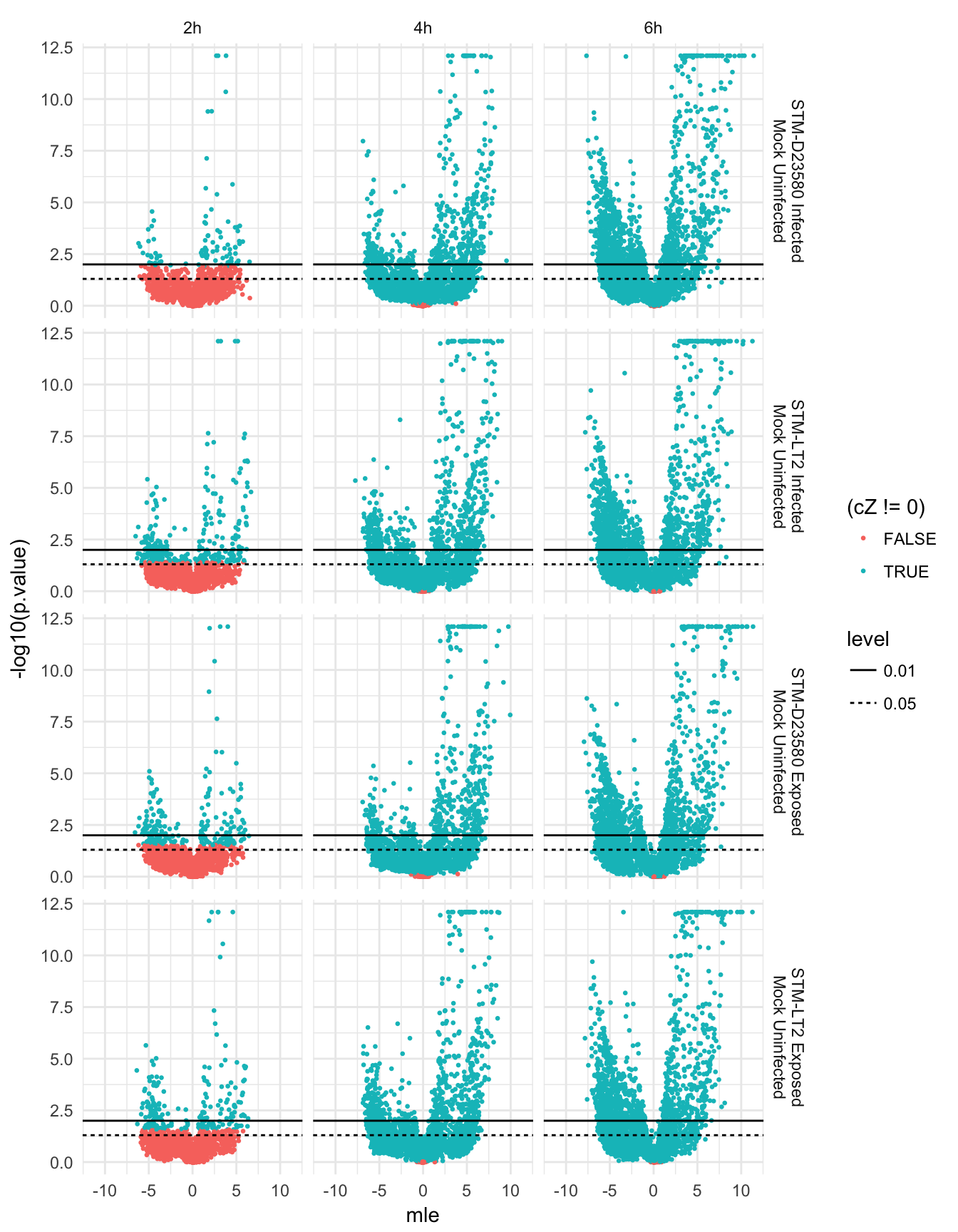
Direct
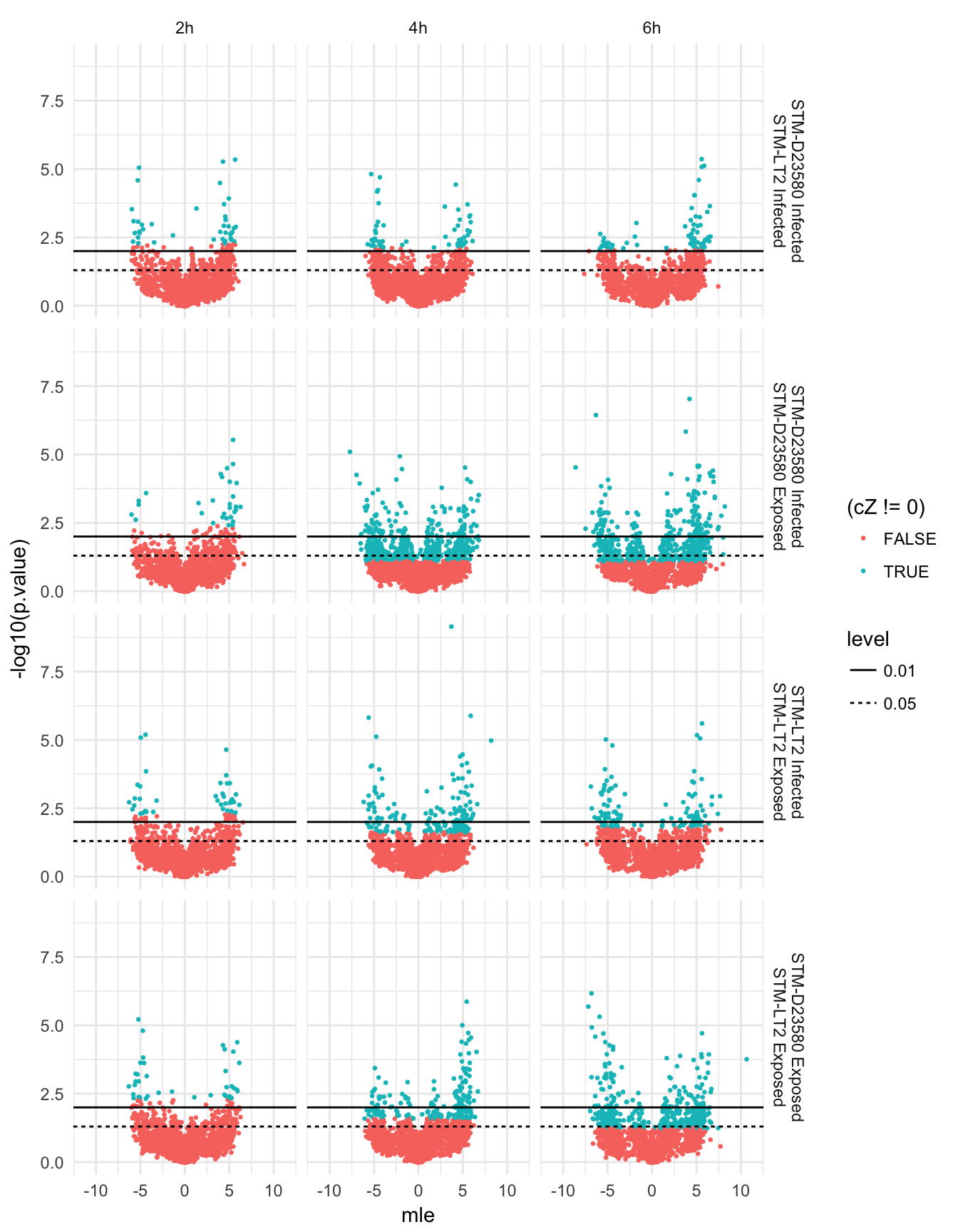
Count DE genes at various cut-offs
Uninfected
| P.01 | P.05 | cZ | |
|---|---|---|---|
| 2h_STM-D23580_Infected-2h_Mock_Uninfected | 84 | 259 | 92 |
| 2h_STM-LT2_Infected-2h_Mock_Uninfected | 136 | 354 | 301 |
| 2h_STM-D23580_Exposed-2h_Mock_Uninfected | 125 | 317 | 229 |
| 2h_STM-LT2_Exposed-2h_Mock_Uninfected | 134 | 328 | 209 |
| 4h_STM-D23580_Infected-4h_Mock_Uninfected | 479 | 927 | 5758 |
| 4h_STM-LT2_Infected-4h_Mock_Uninfected | 618 | 1135 | 7025 |
| 4h_STM-D23580_Exposed-4h_Mock_Uninfected | 443 | 868 | 5547 |
| 4h_STM-LT2_Exposed-4h_Mock_Uninfected | 558 | 1006 | 6696 |
| 6h_STM-D23580_Infected-6h_Mock_Uninfected | 1155 | 1954 | 7338 |
| 6h_STM-LT2_Infected-6h_Mock_Uninfected | 1154 | 1978 | 7348 |
| 6h_STM-D23580_Exposed-6h_Mock_Uninfected | 978 | 1740 | 7274 |
| 6h_STM-LT2_Exposed-6h_Mock_Uninfected | 1090 | 1862 | 7329 |
Direct
| P.01 | P.05 | cZ | |
|---|---|---|---|
| 2h_STM-D23580_Infected-2h_STM-LT2_Infected | 60 | 224 | 42 |
| 2h_STM-D23580_Infected-2h_STM-D23580_Exposed | 55 | 241 | 30 |
| 2h_STM-LT2_Infected-2h_STM-LT2_Exposed | 63 | 210 | 39 |
| 2h_STM-D23580_Exposed-2h_STM-LT2_Exposed | 56 | 220 | 38 |
| 4h_STM-D23580_Infected-4h_STM-LT2_Infected | 64 | 257 | 56 |
| 4h_STM-D23580_Infected-4h_STM-D23580_Exposed | 121 | 401 | 569 |
| 4h_STM-LT2_Infected-4h_STM-LT2_Exposed | 96 | 286 | 178 |
| 4h_STM-D23580_Exposed-4h_STM-LT2_Exposed | 92 | 315 | 190 |
| 6h_STM-D23580_Infected-6h_STM-LT2_Infected | 74 | 279 | 65 |
| 6h_STM-D23580_Infected-6h_STM-D23580_Exposed | 151 | 425 | 641 |
| 6h_STM-LT2_Infected-6h_STM-LT2_Exposed | 85 | 306 | 116 |
| 6h_STM-D23580_Exposed-6h_STM-LT2_Exposed | 109 | 394 | 441 |
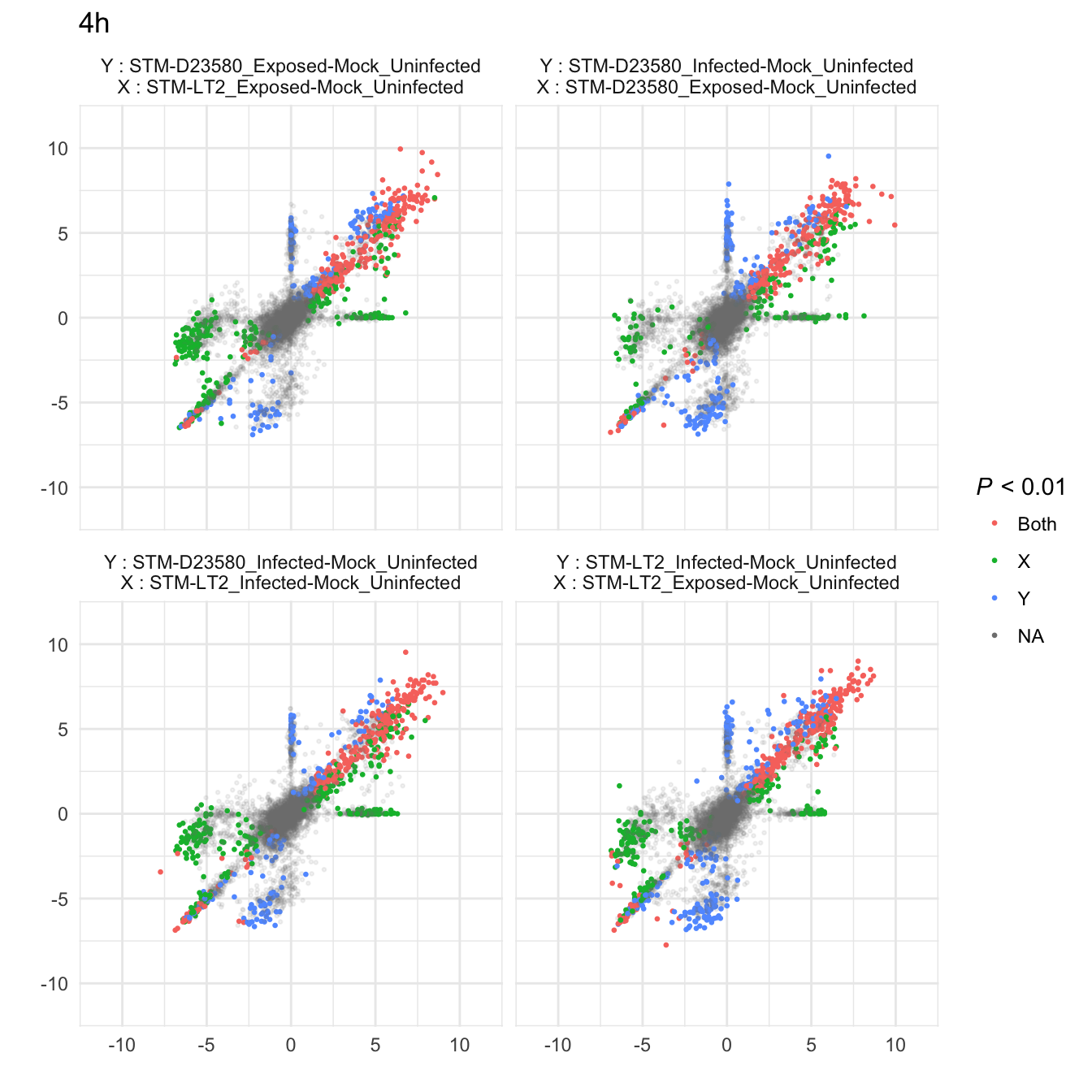
Comparison of fold-change estimates
Contrasts
Let us define:
- the required columns in the scde result tables
resCols <- c("mle","Z")- a function used to annotate each gene in the tables of scde results with the significance of differential expression in the pair of contrasts compared:
add.sig.XY <- function(x, cutoff = 0.01){
sig.levels <- c(NA_character_, "X", "Y", "Both")
alpha.levels <- c(0.3, 1, 1, 1)
sig.x <- (x$p.value.x < cutoff); sig.y <- (x$p.value.y < cutoff)
x$p.01 <- sig.levels[1 + sig.x + 2 * sig.y]
x$alpha <- alpha.levels[1 + sig.x + 2 * sig.y]
return(x)
}- a function used to annotate each gene in the tables of scde results whether it is significantly differentially expressed in opposite direction in the pair of contrasts compared:
addOpposite <- function(x, cutoff = 0.01){
x$opposite <- NA_character_
x.sig.idx <- (x$p.value.x < cutoff & x$p.value.y < cutoff)
x.opposite <- (x$mle.x * x$mle.y) < 0
x[x.sig.idx, "opposite"] <- x.opposite[x.sig.idx]
return(x)
}- the list of contrasts to contrast:
contrasts.contrasts.2h <- list(
c("2h_STM-D23580_Infected-2h_Mock_Uninfected","2h_STM-LT2_Infected-2h_Mock_Uninfected"),
c("2h_STM-D23580_Infected-2h_Mock_Uninfected","2h_STM-D23580_Exposed-2h_Mock_Uninfected"),
c("2h_STM-LT2_Infected-2h_Mock_Uninfected","2h_STM-LT2_Exposed-2h_Mock_Uninfected"),
c("2h_STM-D23580_Exposed-2h_Mock_Uninfected","2h_STM-LT2_Exposed-2h_Mock_Uninfected")
)
contrasts.contrasts.4h <- list(
c("4h_STM-D23580_Infected-4h_Mock_Uninfected","4h_STM-LT2_Infected-4h_Mock_Uninfected"),
c("4h_STM-D23580_Infected-4h_Mock_Uninfected","4h_STM-D23580_Exposed-4h_Mock_Uninfected"),
c("4h_STM-LT2_Infected-4h_Mock_Uninfected","4h_STM-LT2_Exposed-4h_Mock_Uninfected"),
c("4h_STM-D23580_Exposed-4h_Mock_Uninfected","4h_STM-LT2_Exposed-4h_Mock_Uninfected")
)
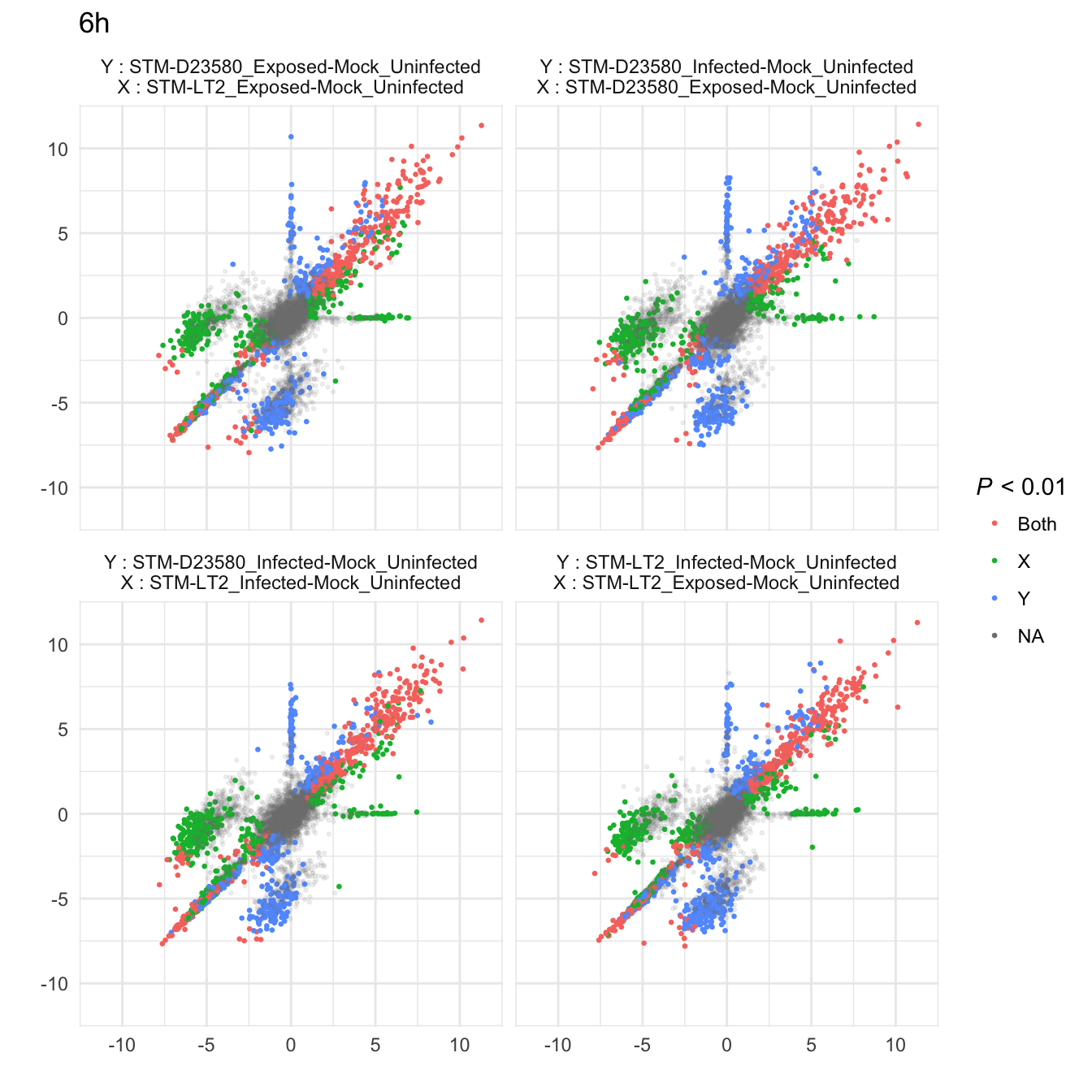
contrasts.contrasts.6h <- list(
c("6h_STM-D23580_Infected-6h_Mock_Uninfected","6h_STM-LT2_Infected-6h_Mock_Uninfected"),
c("6h_STM-D23580_Infected-6h_Mock_Uninfected","6h_STM-D23580_Exposed-6h_Mock_Uninfected"),
c("6h_STM-LT2_Infected-6h_Mock_Uninfected","6h_STM-LT2_Exposed-6h_Mock_Uninfected"),
c("6h_STM-D23580_Exposed-6h_Mock_Uninfected","6h_STM-LT2_Exposed-6h_Mock_Uninfected")
)2h
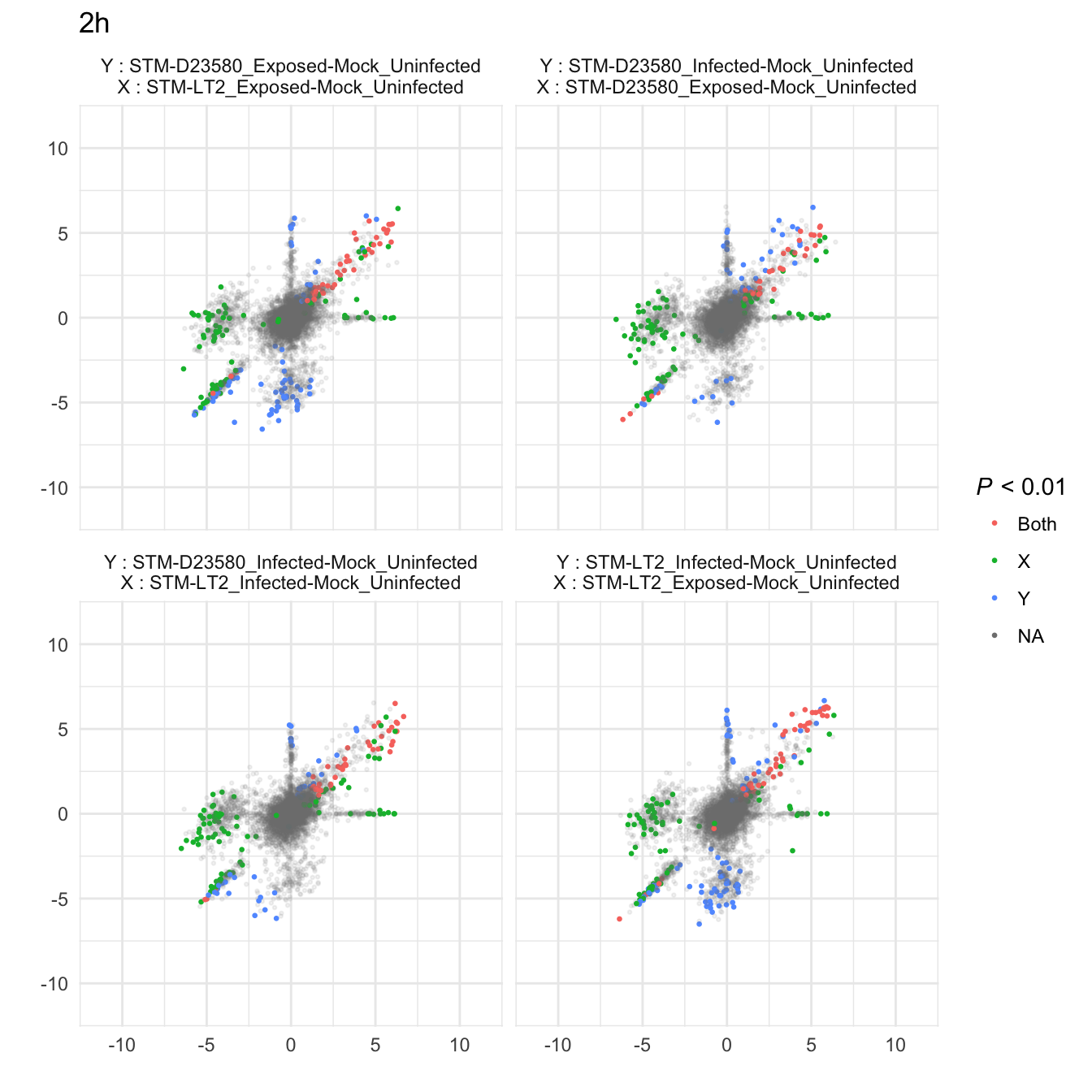
4h
6h