Chapter 1 Plotting reduced dimensions
1.1 Foreword
When one thinks of single-cell data analysis, one thinks of \(t\)-SNEs. (Or maybe UMAPs, for the younger folks.) Indeed, hardly a single-cell paper comes out these days without a \(t\)-SNE or UMAP plot in it somewhere. I would guess that their popularity stems from the entrancing illusion that a viewer is looking at “raw data”, unsullied by the authors’ interpretation. Indeed, I personally prefer \(t\)-SNEs as - with the right parameters - they can be made to look like histology images, while UMAPs look more like the aftermath of blowing your nose. But hey, to each their own.
1.2 Setting up the data
Here we’ll use one of the many PBMC datasets generated by 10X Genomics (Zheng et al. 2017). One can only imagine how many times these PBMCs have been re-analyzed, surely there’s nothing we don’t know about these cells anymore.
library(DropletTestFiles)
fpath <- getTestFile(file.path("tenx-3.0.0-pbmc_10k_protein_v3",
"1.0.0/filtered.tar.gz"), prefix=TRUE)
tmp <- tempfile()
untar(fpath, exdir=tmp)
library(DropletUtils)
sce <- read10xCounts(file.path(tmp, "filtered_feature_bc_matrix"))
sce <- splitAltExps(sce, rowData(sce)$Type) # splitting off the ADTs.
sce## class: SingleCellExperiment
## dim: 33538 7865
## metadata(1): Samples
## assays(1): counts
## rownames(33538): ENSG00000243485 ENSG00000237613 ... ENSG00000277475
## ENSG00000268674
## rowData names(3): ID Symbol Type
## colnames: NULL
## colData names(2): Sample Barcode
## reducedDimNames(0):
## altExpNames(1): Antibody CaptureWe slap together a quick-and-dirty analysis with some of the usual packages (Amezquita et al. 2020). Normally we would make some more diagnostic plots for each of these steps, but it would not do to distract from the star of the show in this chapter.
library(scater)
library(scran)
library(scuttle)
library(bluster)
# Quality control on the mitochondrial content:
qcstats <- perCellQCMetrics(sce, subsets=list(Mito=grep("^MT-", rowData(sce)$Symbol)))
discard <- isOutlier(qcstats$subsets_Mito_percent, type="higher")
sce <- sce[,!discard]
summary(discard)## Mode FALSE TRUE
## logical 7569 296# Normalization and feature selection:
sce <- logNormCounts(sce)
dec <- modelGeneVarByPoisson(sce)
hvgs <- getTopHVGs(dec, n=4000)
# PCA and some clustering:
set.seed(117)
sce <- runPCA(sce, subset_row=hvgs)
colLabels(sce) <- clusterRows(reducedDim(sce), NNGraphParam())
# Creating those sweet, sweet t-SNEs.
set.seed(118)
sce <- runTSNE(sce, dimred="PCA")
sce <- runUMAP(sce, dimred="PCA")
sce## class: SingleCellExperiment
## dim: 33538 7569
## metadata(1): Samples
## assays(2): counts logcounts
## rownames(33538): ENSG00000243485 ENSG00000237613 ... ENSG00000277475
## ENSG00000268674
## rowData names(3): ID Symbol Type
## colnames: NULL
## colData names(4): Sample Barcode sizeFactor label
## reducedDimNames(3): PCA TSNE UMAP
## altExpNames(1): Antibody CaptureWe can see that the dimensionality reduction results are tucked away in the reducedDims() of the sce object.
If you want to get in on the action, you can pull out the reduced dimensions yourself for plotting:
## [,1] [,2]
## [1,] 6.408 -22.65
## [2,] 5.258 -20.71
## [3,] 1.808 -20.09
## [4,] 8.337 25.32
## [5,] 5.944 29.59
## [6,] -5.257 -31.471.3 Visualizing with scater
We’ll start with the visualizations from the scater package (McCarthy et al. 2017).
The plotTSNE() function creates ggplot objects with various aesthetics, as shown in Figure 1.1.
Here, we have coloured each cell by its assigned label, throwing in the label name for good measure.
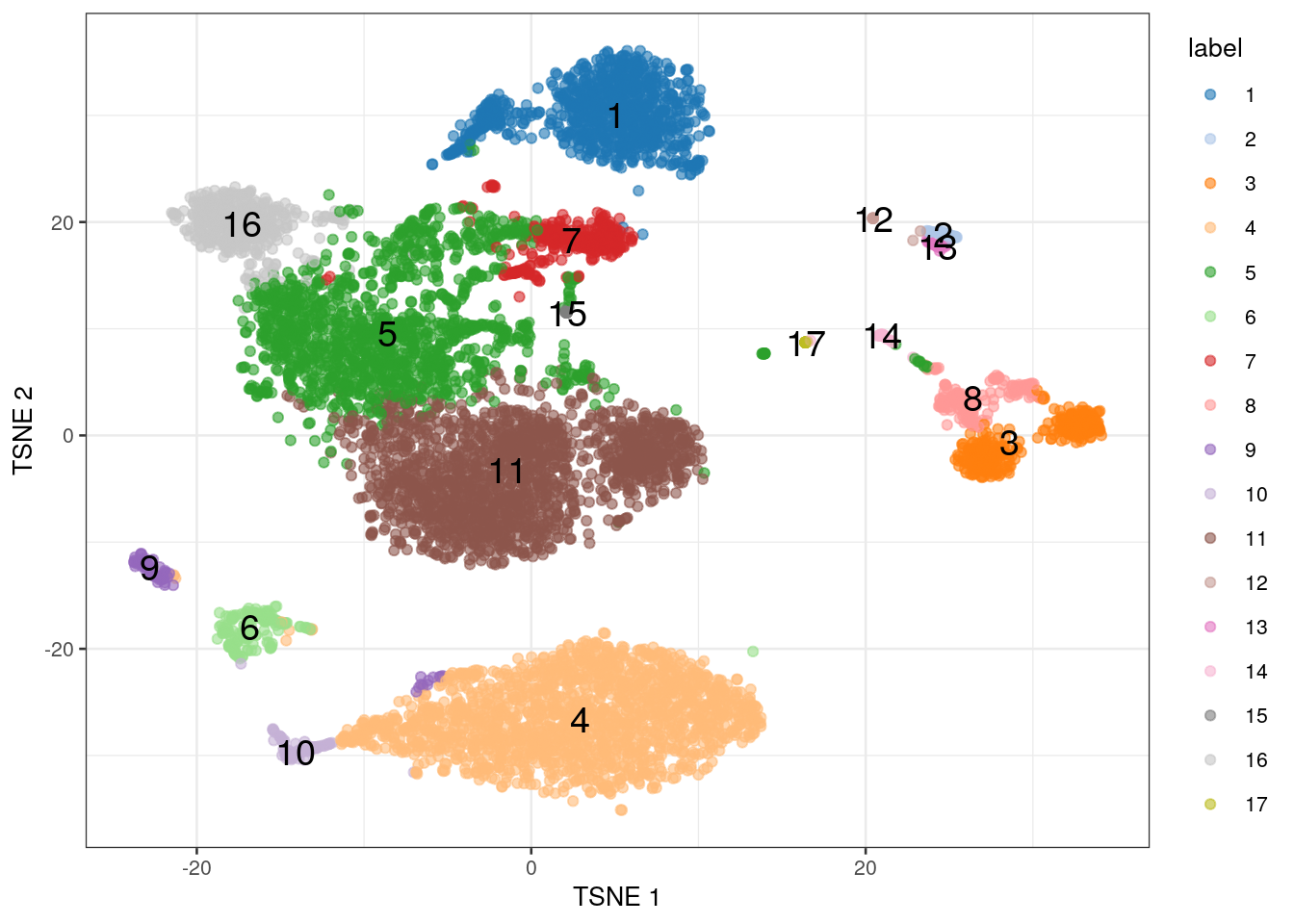
Figure 1.1: \(t\)-SNE coloured by the cluster label.
Alternatively, we might color each cell by the expression of a particular gene,
using the neon 80’s-throwback color scheme that is viridis (Figure 1.2).
We set swap_rownames= to allow us to refer to genes using symbols rather than the more obtuse Ensembl identifiers.
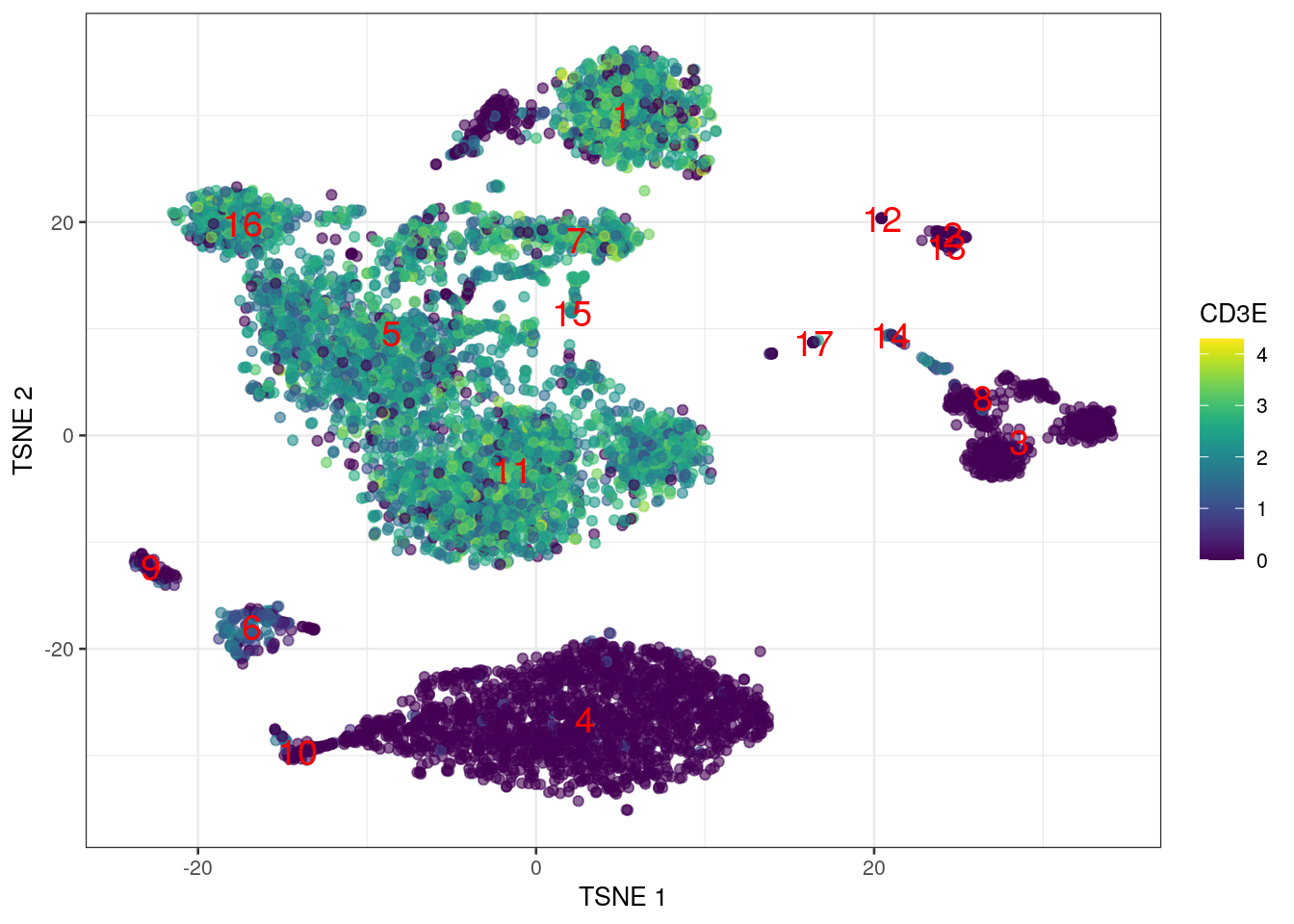
Figure 1.2: \(t\)-SNE coloured by the expression of CD3E.
This function (and its sister plotUMAP()) use the same plotReducedDim() function under the hood.
We can generate plots for any dimensionality reduction result in sce by calling plotReducedDim() directly,
passing in the name of the result that we wish to visualize (Figure 1.3).
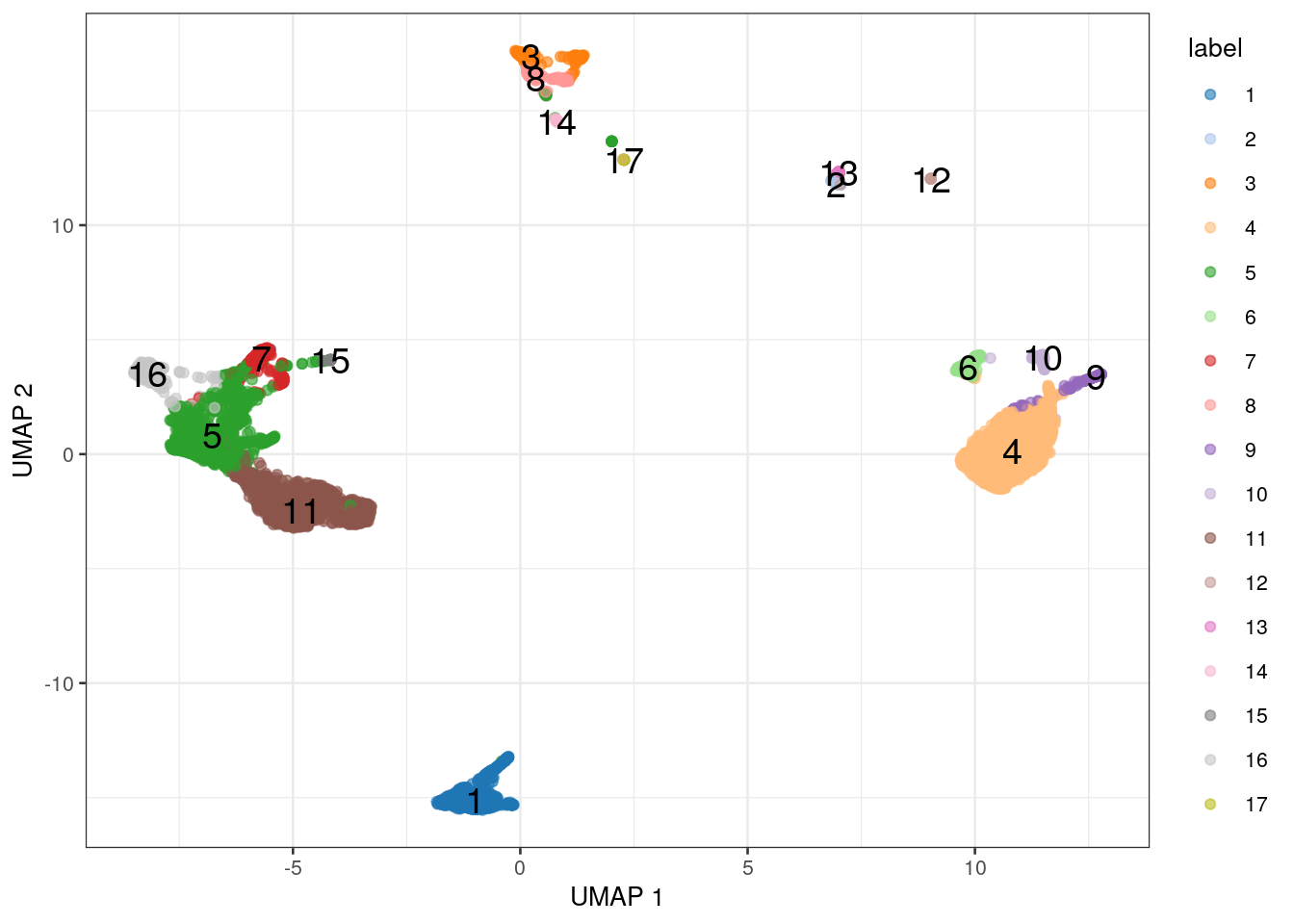
Figure 1.3: UMAP coloured by the cluster label.
And of course, this is all ggplot-based, so you can just add on your own layers to customize the aesthetics.
Don’t like viridis?
(Does anybody? Ho ho ho.)
You can swap it out with a color scale of your choice using the relevant ggplot2 functions,
as shown in Figure 1.4 with a grey-orange gradient for CD3E expression.
plotUMAP(sce, colour_by="CD3E", swap_rownames="Symbol") +
scale_color_gradient(low="grey", high="orange")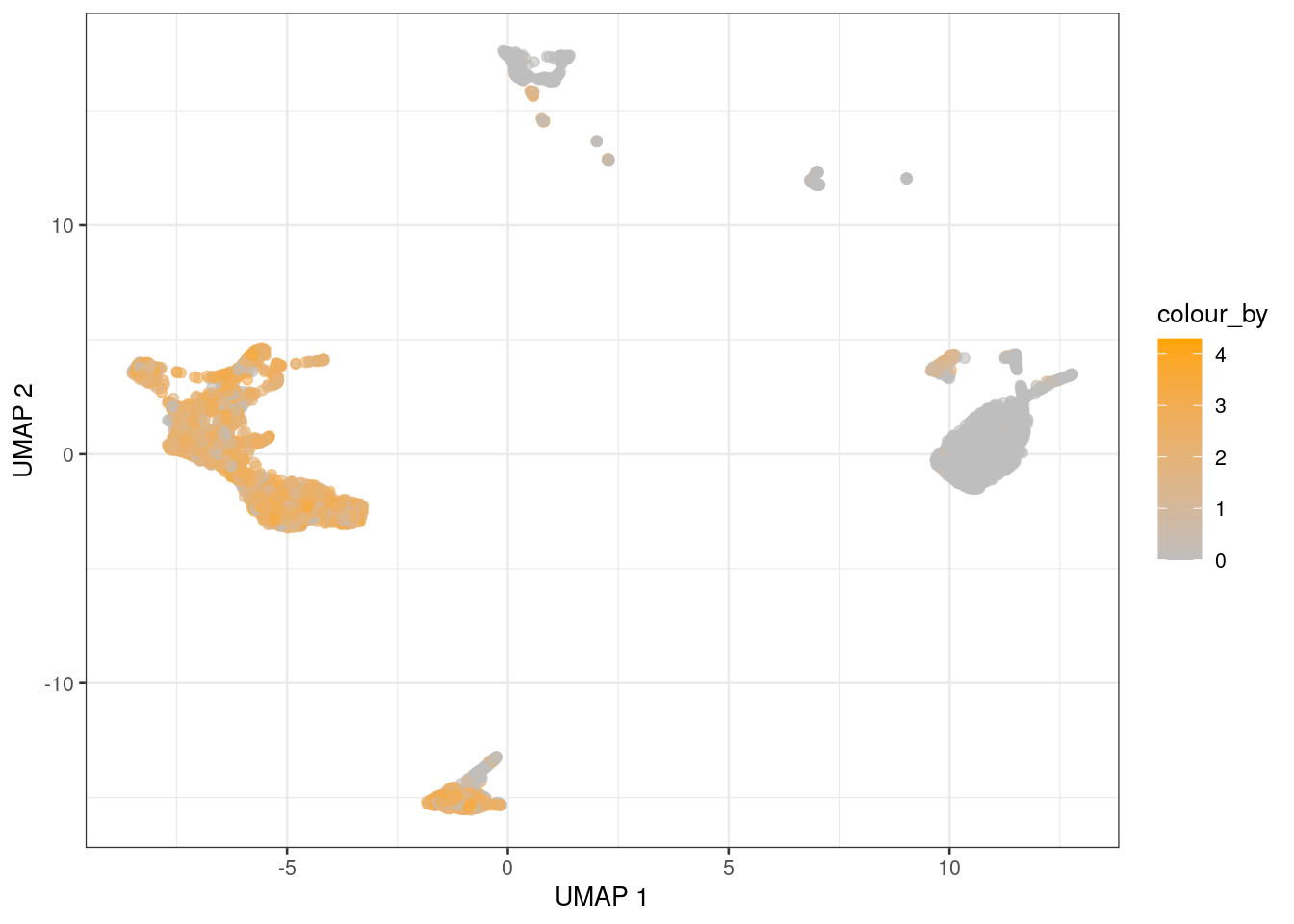
Figure 1.4: UMAP coloured by the expression of CD3E.
In fact, you can tell plotTSNE() and plotUMAP() to carry over any colData() field from the SingleCellExperiment.
These fields are inserted into the data.frame and can be used in any ggplot2-compatible function.
In Figure 1.5, we bin each cell based on its size factor to create the category field,
and we use those bins for faceting the plot generated by plotTSNE().
sce$category <- cut(log(sizeFactors(sce)), 3)
plotTSNE(sce, colour_by="label", other_fields="category") +
facet_wrap('category')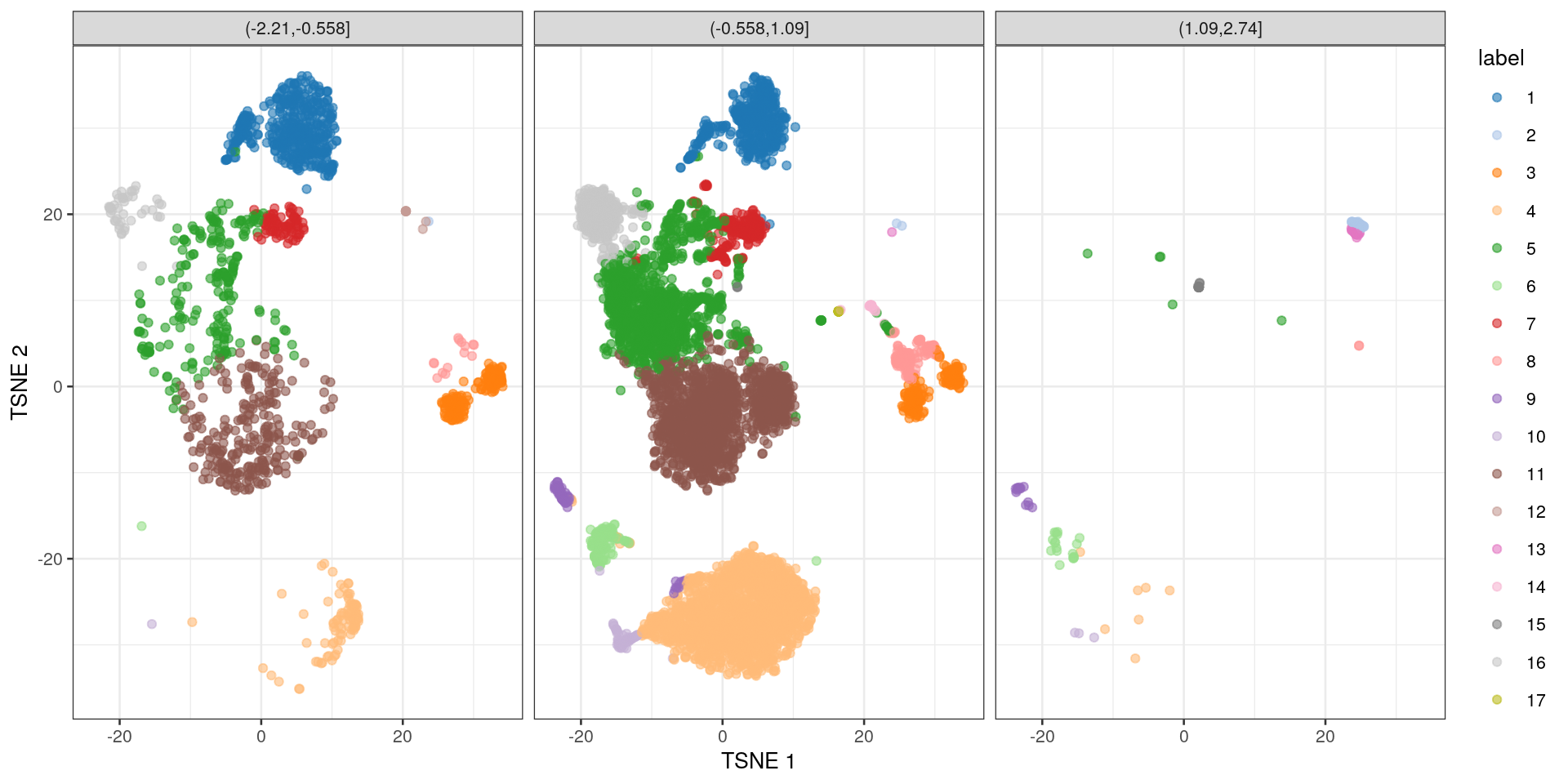
Figure 1.5: \(t\)-SNE coloured by the cluster identity and faceted by the log-size factor after binning.
While most customization can be performed by adding further ggplot-related layers,
we can also tinker with other aesthetic parameters in the plotTSNE() call.
For example, we change the point size and axis labels while also removing the legend in Figure ??.
Further options can be found by following the breadcrumbs in the documentation for ?plotTSNE.

I could go on, but I won’t.
Session information
R version 4.0.2 Patched (2020-09-10 r79182)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.1 LTS
Matrix products: default
BLAS/LAPACK: /usr/lib/x86_64-linux-gnu/openblas-openmp/libopenblasp-r0.3.8.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=C
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] parallel stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] bluster_0.99.1 scuttle_0.99.13
[3] scran_1.17.16 scater_1.17.4
[5] ggplot2_3.3.2 DropletUtils_1.9.11
[7] SingleCellExperiment_1.11.6 SummarizedExperiment_1.19.6
[9] DelayedArray_0.15.7 matrixStats_0.56.0
[11] Matrix_1.2-18 Biobase_2.49.1
[13] GenomicRanges_1.41.6 GenomeInfoDb_1.25.11
[15] IRanges_2.23.10 S4Vectors_0.27.12
[17] BiocGenerics_0.35.4 DropletTestFiles_0.99.5
[19] BiocStyle_2.17.0 BiocManager_1.30.10
loaded via a namespace (and not attached):
[1] Rtsne_0.15 ggbeeswarm_0.6.0
[3] colorspace_1.4-1 ellipsis_0.3.1
[5] XVector_0.29.3 BiocNeighbors_1.7.0
[7] farver_2.0.3 bit64_4.0.5
[9] RSpectra_0.16-0 interactiveDisplayBase_1.27.5
[11] AnnotationDbi_1.51.3 codetools_0.2-16
[13] R.methodsS3_1.8.1 knitr_1.29
[15] dbplyr_1.4.4 R.oo_1.24.0
[17] rebook_0.99.5 graph_1.67.1
[19] uwot_0.1.8 shiny_1.5.0
[21] HDF5Array_1.17.3 compiler_4.0.2
[23] httr_1.4.2 dqrng_0.2.1
[25] assertthat_0.2.1 fastmap_1.0.1
[27] limma_3.45.14 later_1.1.0.1
[29] BiocSingular_1.5.0 htmltools_0.5.0
[31] tools_4.0.2 rsvd_1.0.3
[33] igraph_1.2.5 gtable_0.3.0
[35] glue_1.4.2 GenomeInfoDbData_1.2.3
[37] dplyr_1.0.2 rappdirs_0.3.1
[39] Rcpp_1.0.5 vctrs_0.3.4
[41] rhdf5filters_1.1.2 ExperimentHub_1.15.3
[43] DelayedMatrixStats_1.11.1 xfun_0.17
[45] stringr_1.4.0 ps_1.3.4
[47] mime_0.9 lifecycle_0.2.0
[49] irlba_2.3.3 statmod_1.4.34
[51] XML_3.99-0.5 AnnotationHub_2.21.5
[53] edgeR_3.31.4 zlibbioc_1.35.0
[55] scales_1.1.1 promises_1.1.1
[57] rhdf5_2.33.7 yaml_2.2.1
[59] curl_4.3 memoise_1.1.0
[61] gridExtra_2.3 stringi_1.5.3
[63] RSQLite_2.2.0 highr_0.8
[65] BiocVersion_3.12.0 BiocParallel_1.23.2
[67] rlang_0.4.7 pkgconfig_2.0.3
[69] bitops_1.0-6 evaluate_0.14
[71] lattice_0.20-41 purrr_0.3.4
[73] Rhdf5lib_1.11.3 labeling_0.3
[75] CodeDepends_0.6.5 bit_4.0.4
[77] processx_3.4.4 tidyselect_1.1.0
[79] RcppAnnoy_0.0.16 magrittr_1.5
[81] bookdown_0.20 R6_2.4.1
[83] generics_0.0.2 DBI_1.1.0
[85] pillar_1.4.6 withr_2.2.0
[87] RCurl_1.98-1.2 tibble_3.0.3
[89] crayon_1.3.4 BiocFileCache_1.13.1
[91] rmarkdown_2.3 viridis_0.5.1
[93] locfit_1.5-9.4 grid_4.0.2
[95] blob_1.2.1 callr_3.4.4
[97] digest_0.6.25 xtable_1.8-4
[99] httpuv_1.5.4 R.utils_2.10.1
[101] munsell_0.5.0 beeswarm_0.2.3
[103] viridisLite_0.3.0 vipor_0.4.5 Bibliography
Amezquita, R. A., A. T. L. Lun, E. Becht, V. J. Carey, L. N. Carpp, L. Geistlinger, F. Marini, et al. 2020. “Orchestrating single-cell analysis with Bioconductor.” Nat. Methods 17 (2): 137–45.
McCarthy, D. J., K. R. Campbell, A. T. Lun, and Q. F. Wills. 2017. “Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R.” Bioinformatics 33 (8): 1179–86.
Zheng, G. X., J. M. Terry, P. Belgrader, P. Ryvkin, Z. W. Bent, R. Wilson, S. B. Ziraldo, et al. 2017. “Massively parallel digital transcriptional profiling of single cells.” Nat Commun 8 (January): 14049.